Blood provides a mechanism by which nutrients, gases, and wastes can be transported throughout the body. It consists of a number of cells suspended in a fluid medium known as plasma. Serum refers to plasma after clotting factors and fibrin have been removed.
Peripheral Blood Smear
The cells of the blood are important because they are a readily accessible population whose morphology, biochemistry, and ecology may give indications of a patient's general state or clues to the diagnosis of disease. For this reason, the complete blood count (CBC) and the white blood cell (WBC) differential are routinely used in clinical medicine. It is very important to be able to recognize normal blood cells and to distinguish pathological cells from the normal variants.
The identification of blood elements is based primarily on observations of the presence or absence of a nucleus and cytoplasmic granules. Other helpful features are cell size, nuclear size and shape, chromatin appearance, and cytoplasmic staining.
Component Cells of the Blood Smear
A blood smear is created by placing a drop of blood near the end of a clean glass microscope slide. Another slide is held at an angle, backed into the drop, and then smoothly dragged forward to spread the blood film along the slide. The blood must then be fixed, stained, and washed.
When you view a properly prepared blood smear of a healthy individual, there are several populations of cells that you will notice.
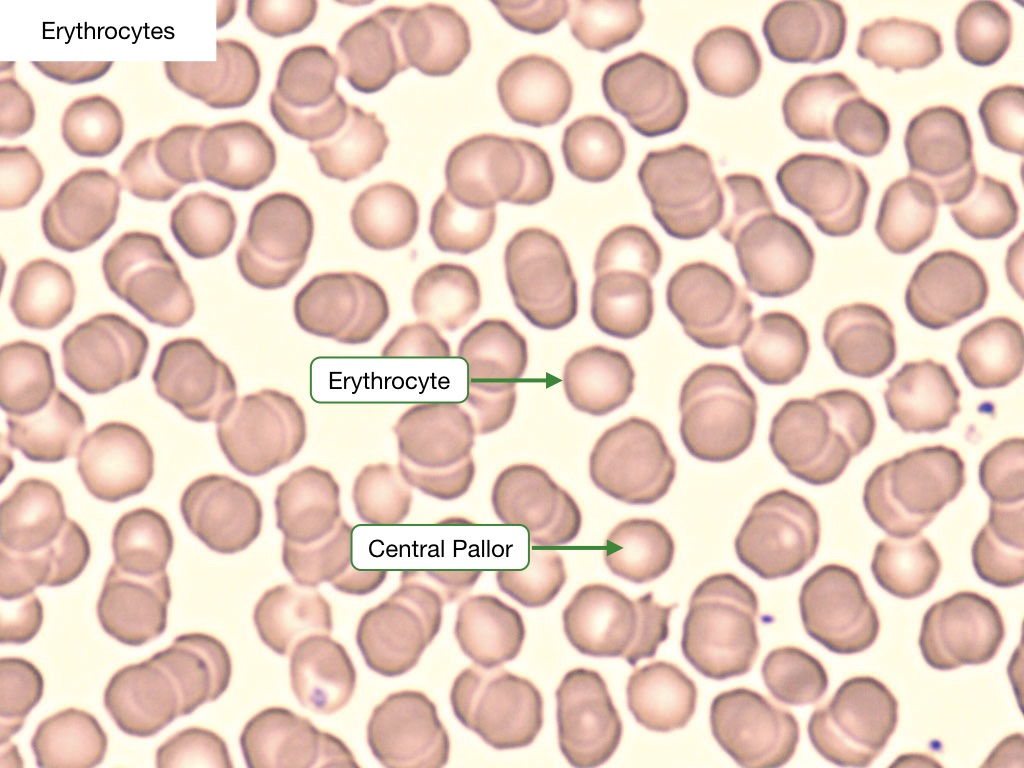
Erythrocytes
Erythrocytes, or red blood cells (RBC), are by far the predominant cell type in the blood smear. They are anucleate, non-granulated, eosinophilic cells that are uniform in shape (biconcave discs) and size (7.2 microns). RBCs have a central cavity (a.k.a., central pallor) that appears pale under the light microscope. These cells contain hemoglobin and are responsible for the transport and delivery of oxygen. RBCs have a lifespan of 120 days. The size of an RBC is measured automatically when a complete blood count (CBC) is performed; this is represented by the mean corpuscular volume (MCV), with normal being ~80-100 fL. Another useful RBC parameter measured on the CBC is red cell distribution width (RDW), which describes the size distribution of the entire RBC population, with normal being defined as less than 15%.
Reticulocytes
Reticulocytes are immature RBCs that are released from the bone marrow. They mature into RBCs after 1 to 2 days in the peripheral blood. There should be about one reticulocyte for every 100 red blood cells in a normal blood smear. These cells stain with a light blue tint because they still have RNA-containing organelles such as free ribosomes.
Platelets
Platelets are the smallest elements of the blood and are responsible for the formation of clots through a complex, highly regulated cascade. Platelets are between 2 and 5 microns in diameter and appear ovoid and anucleate with purple granules.
Leukocytes
Leukocytes, or WBCs, are cells of the immune system that are present in both blood and interstitial fluid. There should be about 1 leukocyte for every 1000 RBCs. They can be classified into two groups according to their nuclear pattern and the presence of cytoplasmic granules.
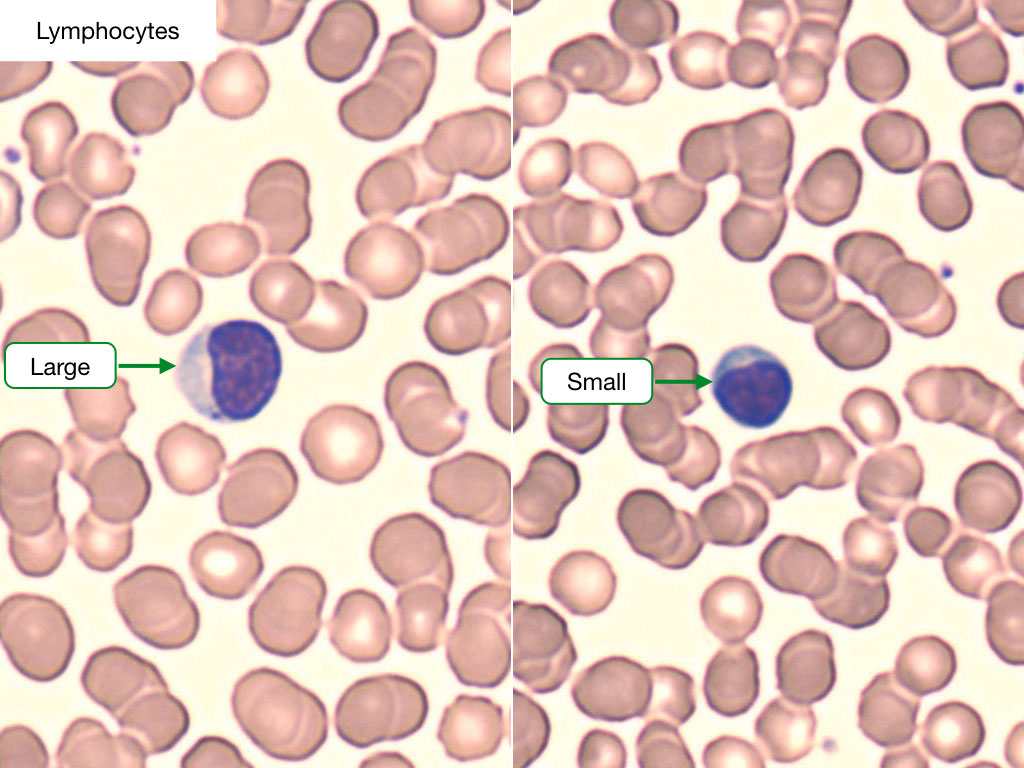
Lymphocytes
Lymphocytes are about the same size as RBCs and have deeply stained nuclei with a thin rim of cytoplasm. The major population of lymphocytes is composed of B-cells and T-cells. A third population, known as natural killer (NK) cells, can also be identified. Lymphocyte counts are raised in response to viral infections.
Monocytes
Monocytes are larger than lymphocytes and have less-clearly demarcated nuclei that are usually not centered in the cell. These nuclei appear horseshoe-shaped, and the cytoplasm contains fine granules that give it a muddy gray color. These granules contain lysosomal enzyme and peroxidase. Monocytes are phagocytic cells that are important in the inflammatory response. They are the precursors to tissue macrophages.
Polymorphonuclear leukocytes
Polymorphonuclear leukocytes are cells with lobated nuclei and cytoplasmic granules. While these cells share the same primary (nonspecific) or azurophilic granules, they are named based upon the characteristics of their secondary (specific) granules.
Neutrophils are by far the most numerous of all WBCs. They are characterized by a nucleus that is segmented into three to five lobes that are joined by slender strands. The cytoplasm of neutrophils stains a pale pink. Its primary granules contain acid hydrolases and cationic proteins, and its secondary granules contain a variety of antimicrobial substances used to destroy bacteria that they phagocytose during the acute inflammatory response.
Eosinophils are larger than neutrophils and are distinguished by large red or orange granules of uniform size. These granules contain major basic protein, which is released to kill organisms too large to phagocytose, such as parasites and helminths.
Basophils are intermediate in size between neutrophils and eosinophils and have simple or bilobed nuclei. They contain many coarse purple granules that can vary in size or shape. These granules contain histamine, which is released to cause a vasoactive response in hypersensitivity reactions, and heparin, which is an anticoagulant. Basophils are not phagocytic.
Transfusion Medicine and Sickle Cell Disease
Introduction
Blood provides a mechanism by which nutrients, gases, and wastes can be transported throughout the body. It consists of a number of cells suspended in a fluid medium known as plasma. Serum refers to plasma after clotting factors and fibrin have been removed.
Hemoglobin Structure
Normal hemoglobin (Hgb) is a complex structure. The primary structure contains amino acids strung together to make a polypeptide globin chain. These globin chains are arranged into helices, forming the secondary structure. The tertiary structure consists of coiled globin chains forming pockets that bind heme; each heme molecule, containing one ferrous iron (Fe2+) molecule, is synthesized in the mitochondria and cytosol of erythroid precursors. Four globin chains then bind to form the final quarternary structure of Hgb, which is a tetramer.
Structure of Hemoglobin
Sickle Cell
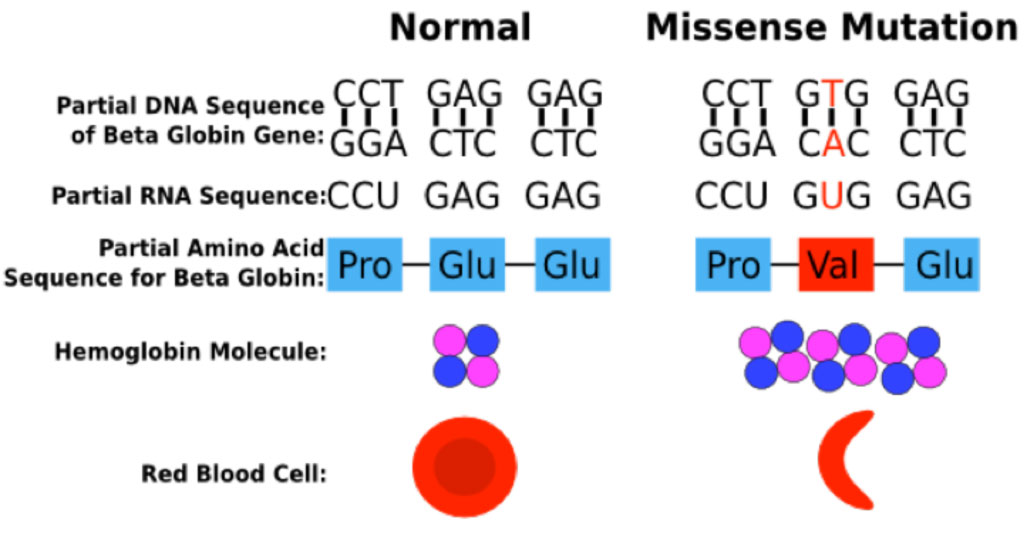
Hemoglobin abnormalities arise when there is a mutation or deletion involving a portion of one of the globin genes. Sickle hemoglobin (Hgb S) results from a point mutation in the beta globin gene at position 6, in which the normal glutamate is replaced by valine.
Sickle Cell Mutation
The Hgb S mutation generates a Hgb molecule with reduced solubility upon deoxygenation. Insoluble Hgb S polymerizes within circulating red blood cells. The polymerization of Hgb S causes dehydration of the red blood cell, leading to a dense intracellular concentration of Hgb S polymers, further enhancing Hgb S polymerization. These changes lead to a distortion in the shape of the red blood cell and a decrease in cell deformability, ultimately resulting in sickling of the red cell.
Polymerization of Hgb S
As red cells traverse the microcirculation, oxygenated hemoglobin releases its oxygen, generating deoxygenated hemoglobin. In patients with Hgb S, the deoxygenated form of Hgb S bears a conformational change in which the β subunits move away from each other, and a hydrophobic patch at the site of the β6valine replacement binds to a complementary hydrophobic site on a β subunit of another hemoglobin tetramer, forming a Hgb S polymer. As deoxygenated Hgb S polymerizes, the red cell is distorted into an elongated banana or "sickle" shape.
Diagnosing Sickle Cell Disease
The laboratory diagnosis of sickle cell disease relies upon hemoglobin electrophoresis to identify the presence of an abnormal Hgb S protein. The hemoglobin electrophoresis in patients with homozygous Hgb S disease typically shows 90-95% Hgb S, 4-5% hemoglobin A2 (Hgb A2), and 0% hemoglobin A (Hgb A).
Electrophoresis of Hemoglobin in Sickle Cell Disease
By contrast, the hemoglobin electrophoresis in patients with Hgb S trait typically shows about 60% Hgb A, 35% Hgb S, and 3-5% Hgb A2.
Electrophoresis of Hemoglobin in Patients with Hgb S Trait
Transfusion to Treat Sickle Cell Disease
Sickle cell trait is generally a clinically silent disorder, while sickle cell disease is associated with a number of complications that arise primarily from vaso-occlusion the microvasculature by sickled red blood cells. Clinical manifestations include acute chest syndrome, stroke, pain crises, leg ulcers, nephropathy, priapism, and pulmonary hypertension. Transfusion of wild-type packed red blood cells (PRBC) can treat or prevent many of the complications of this disorder but carries its own set of complications, including alloimmunization against red cell antigens and transfusional iron overload. Red cell exchange transfusions are indicated in acute chest syndrome but may be logistically difficult to perform as they require placement of specialized intravenous catheters. Hydroxyurea, a chemotherapy agent, has become a mainstay of long-term management of sickle cell disease; it induces increased expression of Hgb F, lessening the extent of deoxygenation within red cells, thereby decreasing the number and severity of pain crises.
Type and Screen Test
Identification of red blood cells for transfusion requires performing a "type and screen". The "typing" process enables determination of ABO blood type and Rh(D) restriction. The patient's red blood cells are incubated with serum known to contain antibodies against A, B, or Rh(D) antigens ("antiserum"); red blood cell agglutination to a specific antiserum indicates that the red cells bear that antigen.
Type and Screen Test
Antibody Screen Test
The "antibody screen" evaluates for the presence of antibodies in the patient's serum. Antibody screening is essentially an indirect Coombs test in which a patient's plasma is incubated with a panel of reagent red blood cells expressing various antigen combinations. Should the plasma contain an antibody against one of these antigens, agglutination will occur. Patients requiring transfusions would then be transfused with PRBC lacking those antigens.
Antibody Screen Test
Direct Anticoagulant Test
The direct anticoagulant test (DAT), or direct Coombs, is a different type of assay that tests for the presence of alloantibodies or autoantibodies located on the red cell surface. A patient's red blood cells that are coated with either IgG or IgM antibodies are incubated with anti-human globulin (AHG) with specificity against IgG or complement protein C3, the latter of which is fixed by IgM. Red cell agglutination in the presence of anti-IgG AHG indicates that the red blood cells are coated with an IgG-fixing antibody; this may be seen as a consequence of hemolytic disease of the fetus and newborn due to maternal-fetal incompatibility to Rh(D), or as a consequence of autoantibodies in a specific type of autoimmune hemolytic anemia (AIHA) known as warm AIHA, which can arise from exposure to certain medications or in autoimmune diseases such as systemic lupus erythematosus. Red cell agglutination in the presence of anti-C3 AHG indicates that the cells are coated with a complement-fixing IgM antibody, which may occur in cold AIHA (as seen in the wake of some infectious such as infectious mononucleosis or Mycoplasma pneumonia (a.k.a. "walking pneumonia")). Note that while warm AIHA is almost always IgG-fixing and can sometimes be C3-fixing as well, cold AIHA is typically only C3-fixing.
Direct Anticoagulant Test
Myeloid Cells
Hematopoiesis
Pluripotent hematopoietic stem cells (HSC) in the marrow differentiate into all of the different circulating blood cells. HSCs commit to the myeloid or lymphoid lineage under the influence of cytokines & growth factors, generating myeloid or lymphoid stem cells. Mature myeloid cells include neutrophils, basophils, eosinophils, and monocytes. Red blood cells (RBC) and platelets are also derived from a common myeloid progenitor but diverge soon thereafter. This process of hematopoiesis is illustrated in the following diagram.
Hematopoiesis
Neutrophil Differentiation
Representative photographs of actual differentiating myeloid cells in the neutrophil lineage are shown here.
Differentiation of Neutrophils
Note that blasts tend to be large cells with a high nucleus-to-cytoplasm ratio, and they often contain large pale circular areas in the nucleus that represent prominent nucleoli. Blasts in the myeloid lineage often are highly granulated (although this characteristic does not always hold).
Blood Cell Count Terminology
In evaluating blood cells, the following terminology is used to describe variations in cell number. Elevations in cell count are labeled with the suffix "-osis" or "-ilia", e.g., leukocytosis, neutrophilia, lymphocytosis, eosinophilia, or thrombocytosis. Reductions in cell count are labeled with the suffix "-penia", e.g., leukopenia, neutropenia, lymphopenia, or thrombocytopenia. An elevation in RBC mass leading to an increased hemoglobin or hematocrit may be referred to as "erythrocytosis" or "polycythemia"; although these terms are often used interchangeably, they are not synonymous, as erythrocytosis refers to an actual increase in RBC mass, while polycythemia refers to any increase in hemoglobin or hematocrit independent of whether or not the RBC mass is high.
Evaluating Blood Cell Counts
In approaching a patient with an elevated blood cell count, the first step is to determine what type of blood cell is affected. The next step is to categorize the elevation in cell count as primary vs. secondary. Primary abnormalities are those that arise from a primary hematologic problem, often due to a bone marrow disorder. Secondary abnormalities are those in which bone marrow function and hematopoiesis are intact, but there is a stimulus exogenous to the marrow that is driving forth increased hematopoietic activity.
As an example, consider a patient with a WBC count of 24,000/µn;L (normal 4-10,000) and the following blood smear.
Blood Smear
The first step in evaluating this patient's leukocytosis is to determine which blood cell type is elevated - in this case, neutrophils. The second step is to determine whether the neutrophilia seen here is due to a primary or secondary cause. Primary causes of neutrophilia arise when there is a primary hematologic or bone marrow problem giving rise to a high neutrophil count. A primary neutrophilia might result from a hematopoietic defect due to clonal overproduction of neutrophils, which can be seen in some types of leukemias or similar disorders known as myeloproliferative neoplasms. By contrast, secondary neutrophilia arises when there is an exogenous stimulus that promotes normal albeit accelerated neutrophil production and release from the bone marrow, or that increases intravascular demargination of neutrophils. The major causes of secondary neutrophilia are infections (which tend to give rise to not only an increase in mature circulating neutrophils, but also a rise in circulating bands, a.k.a. "bandemia") or medications, particularly steroids (which increase neutrophil demargination from the vascular wall) or lithium (which causes increased levels of granulocyte colony stimulating factor (G-CSF)). In general, for most cases of elevated blood counts, secondary causes will be more likely than primary causes.
Here are some additional examples of how to approach elevations in different myeloid cell counts.
A patient with eosinophilia might have incurred this as a result of a primary problem in the bone marrow, or a secondary process such as an allergic reaction, asthma, or a parasitic infection.
A patient with monocytosis might have a primary problem in the bone marrow, or a secondary process such as an infection (e.g., tuberculosis), an autoimmune or inflammatory disease (e.g., sarcoidosis), splenectomy, or a bone marrow that is recovering from a prior aplastic insult (e.g., from recent chemotherapy).
Basophilia is generally considered abnormal and often raises suspicion for a primary problem in the bone marrow, most commonly a myeloproliferative neoplasm such as polycythemia vera or chronic myeloid leukemia.
Lastly, in order to understand some aspects of this lab, and as a preview for the clinicopathologic conferences that will comprise the bulk of the malignant hematology lectures, we should discuss a few key concepts that underlie the myeloid cancers in hematology.
Myelodysplastic syndrome (MDS) is a diagnosis that is based on morphology of blood and bone marrow cells, in conjunction with circulating cell numbers. The diagnostic criteria require that ≥ 10 of cells have dysplasia - which means that the cells "look funny". Many patients with MDS will also have an increase in the number of myeloid blasts in the bone marrow. The major complications of MDS are the consequences of cytopenias (e.g. neutropenia, which can give rise to increased infections; anemia, leading to fatigue and debilitation; and thrombocytopenia, leading to increased bleeding). In addition, MDS is a clonal disorder, meaning that if one analyzes the DNA from the bone marrow of a patient with MDS, an increased representation of certain DNA mutations specific to that patient's MDS will be identified, indicating that the bone marrow cells have largely arisen from a progenitor cell. Because it is a clonal disorder, MDS carries an increased risk of transformation into acute myeloid leukemia, which is defined by the presence of ≥ 20% myeloid blasts in the blood or bone marrow. Interestingly, MDS can sometimes be hallmarked by the presence of circulating myeloid cells known as Pelger-Huet cells, which have a bilobed nucleus with a thin piece of nuclear material connecting the two nuclear lobes, resembling pince nez glasses, as follows:
Pelger-Huet Cells
Note that the original Pelger-Huet anomaly was first described as an inherited autosomal dominant condition sometimes be associated with certain anatomic abnormalities. Hence, most clinicians refer to the Pelger-Huet cells of MDS as "pseudo"-Pelger Huet cells to distinguish them from the true Pelger-Huet anomaly.
Myeloproliferative neoplasms (MPN) are a heterogeneous group of disorders characterized by excessive proliferation of different hematopoietic cell types. There are four MPNs:
Polycythemia vera (PV) is characterized by an increase in hemoglobin and hematocrit with an increase in red cell mass.
Essential thrombocytosis (ET) is characterized by an increase in platelet count. A typical CBC and blood smear in ET will show thrombocytosis, with all other secondary causes of thrombocytosis being ruled out (e.g., infection, inflammation, or iron deficiency).
Primary myelofibrosis (PMF) is characterized by an increase in fibrosis (scar tissue) in the marrow, leading to extramedullary hematopoiesis (i.e., hematopoiesis at sites other than the bone marrow, most commonly the spleen and/or sometimes the liver) and consequent splenomegaly +/- hepatomegaly. A typical CBC and blood smear in PMF will show a large number of early granulocyte forms including blasts and early RBC forms including nucleated red blood cells, referred to as a "leukoerythroblastic" picture, along with red cells that have a teardrop shape (a.k.a. "dacrocytes""), owing to copious scar tissue in the marrow, which limits normal hematopoietic potential and alters the types and morphologies of cells released from the marrow.
Chronic myeloid leukemia (CML) is characterized by an increase in representation of myeloid cells at all stages of myeloid differentiation.
At least two major molecular mutations underlie the different MPNs. PV, ET, and PMF are associated with a mutation in a tyrosine kinase known as the JAK2 V617F mutation, which is seen in about 98% of PV cases and 40-50% of ET and PMF. CML develops because of an abnormal tyrosine kinase known as Bcr-Abl, which arises from a translocation between the abl gene on chromosome 9 and the bcr gene on chromosome 22, a.k.a. the "Philadelphia chromosome", or t(9;22). Treatment of CML was revolutionized a number of years ago by the development of an inhibitor to Bcr-Abl known as imatinib (Gleevec®), which heralded the dawn of the molecular therapeutics in cancer.
Acute myeloid leukemia (AML) is a diagnosis that is based on the finding of more than 20% myeloid blasts in the blood or bone marrow. AML is treated with intensive chemotherapy in many cases, with consideration of hematopoietic stem cell transplant in high-risk patients.
Lymphoid Cells
Types of Lymphoid Cells
There are three major types of lymphoid cells: B-cells, T-cells, and Natural Killer (NK) cells. B-cells develop in the bone marrow and are responsible for production of immunoglobulins (a.k.a. antibodies). T-cells develop in the thymus; these cells have a surface T cell receptor (TCR) that recognizes antigens bound to major histocompatibility complex (MHC) receptors on the surfaces of antigen presenting cells, leading to T-cell activation and various immune system effects. NK cells arise from bone marrow and are stimulated by interferon to recognize and kill "non-self" cells including microorganisms and cancer cells.
Development of B-cells
All lymphocytes descend from a progenitor lymphoid stem cell, which gives rise to a lymphoid blast, which can be recognized on light microscopy by virtue of being larger than a mature lymphocyte and bearing a high nuclear:cytoplasm ratio and prominent nucleoli in the nucleus. B cell development in the bone marrow is highly coordinated, beginning with creation of a pro-B cell and culminating in a mature B cell. A number of different cell surface markers known as Cluster of Differentiation (CD) proteins are produced during B cell development; these CD proteins characterize the different stages of B cell maturation. Also during this time, the immunoglobulin (Ig) genes that will eventually encode the mature antibodies undergo a series of complex gene rearrangements via a process known as V(D)J recombination. The end result is a mature B cell that contains rearranged Ig genes encoding a transmembrane Ig protein on the cell surface that acts as the B cell receptor for foreign antigens. Once the mature B cell becomes activated by binding of the B cell receptor to a specific antigen (as occurs in the setting of an active infection), it differentiates into two types of cells: a plasma cell, which is a terminally-differentiated effector B cell whose major function is to secrete antibodies, and a memory B cell, which serves as a long-term genetic reservoir of immunologic memory in the event of a future infection by the same microbe.
Markers During B-cell Development
Some of the major CD proteins in B cell development are CD19 and CD20, which are produced throughout much of B cell development; CD10, which is a marker of the part of a lymph node known as the germinal center (described further below); and CD38, which is expressed in plasma cells.
Development of T-cells
Like B cell development, T cell development also proceeds via a series of highly complex, regulated steps, beginning with a pro-T cell and ending in a mature T cell. The T cells express a variety of different CD markers at different stages of maturation, most notably CD4 or CD8, which identify two different types of T cells. CD4+ T cells are known as helper T cells as they assist in B cell receptor-antigen interactions. CD8+ T cells are known as cytotoxic T cells as they function to kill other cell types by triggering apoptosis.
Markers During T-cell Development
Some of the major CD proteins in T cell development are CD3, which is present on all T cells; CD4, which identifies CD4+ helper T cells; and CD8, which identifies CD8+ cytotoxic T cells. Clinically, CD4 and CD8 are important in patients with HIV, which infects CD4+ T cells, leading to a reduction in CD4+ to CD8+ T cell ratio (which under normal circumstances is around 2:1).
Histological Appearance of B-cells and T-cells
B and T lymphocytes appear identical under light microscopy. Each is ~7-15 µn;m, with blue cytoplasm and an ovoid nucleus containing coarse chromatin. A representative lymphocyte is shown below.
Lymphocyte
NK Cells
Whereas B and T cells are part of the adaptive immune system (i.e., the immune system that governs antigen-specific immune responses), NK cells are part of the innate immune system in that they serve to provide general, rather than antigen-specific, immunity. They express CD16 and CD56 on their cell surfaces and are negative for the T cell marker CD3. Morphologically they are large, unusual-appearing lymphocytes, a representative of which is shown below.
NK Cells
Identification of Lymphoid Cells by Flow Cytometry
T-cells, B-cells, and NK-cells can also be differentiated from one another by using flow cytometry to identify their proteins and cell surface markers. In this technique, cells are stained with fluoroscopic markers that each have specificity against a different cellular protein. The stained sample is then run through a machine that exposes the cells to a laser; any cells bearing the specific protein(s) in question will fluoresce, and the different wavelengths at which the cells fluoresce allow for identification of multiple different cell types. Consider, for example, the following flow cytometry plot
Flow Cytometry Plot of CD5 and CD 19
This shows one population of cells that express CD19, a pan-B cell marker, plus CD5, a T cell marker, in addition to a smaller population of cells that are CD5+ CD19-.
Lymphoid System
A basic understanding of the lymphoid system is essential in order to understand the various lymphocyte disorders. The lymphoid system contains primary and secondary lymphoid organs. Primary lymphoid organs include the bone marrow, where B cell development occurs, and the thymus, the site of T cell development. Secondary lymphoid organs include the lymph nodes, mucosa-associated lymphoid tissue (MALT), and the spleen. The secondary lymphoid organs are the sites where antigenic stimulation of B cells occurs. There are hundreds of lymph nodes disseminated all over the body. Each has a defined structure, consisting of primary lymphoid follicles where mature non-activated B cell congregate, and secondary lymphoid follicles (a.k.a. germinal centers), which arise when B cells in primary follicles become activated by antigenic stimulation. A schematic of a typical lymph node is shown here.
Cartoon Lymph Node
The orderly architecture of the lymph node often (but not always) becomes disrupted if the lymph node is infiltrated by a malignancy of the lymphoid cells. Such malignancies may be categorized as leukemia or lymphoma. Historically, leukemia was invoked to describe a condition in which the malignant cells were presently primarily in the blood and bone marrow, while lymphoma was used to describe a lymphoid malignancy where the malignant cells primarily homed to the lymph nodes, spleen, and peripheral lymphoid tissues. We now know that this is an oversimplification, since many patients with lymphoma will have malignant cells in the blood and/or bone marrow, and many with leukemia will have lymph node and splenic involvement - a classic example being chronic lymphocytic leukemia (CLL), an indolent B cell disorder that can sometimes be present exclusively in the blood and bone marrow (where it is referred to as CLL) and other times be present largely in the lymph nodes (where it is referred to as small lymphocytic lymphoma (SLL)). The World Health Organization (WHO) circumvents this problem by categorizing the lymphoid malignancies according to the point at which lymphoid cell development becomes aberrant during lymphoid development, leading to malignancy. According to this designation, most acute lymphoid leukemias such as acute lymphoblastic leukemia will have a defect in early lymphoid differentiation, while most lymphomas will have a defect in mature lymphoid cells.
CLL and SLL are categorized together as a mature B cell malignancy in the WHO classification. Acute lymphoid or acute lymphoblastic leukemia (ALL) is the prototypical lymphoid leukemia resulting from a defect in early B or T cell development. ALL is characterized by the presence of lymphoid blasts, which, like myeloid blasts, are large cells with a high nucleus-to-cytoplasm ratio and, often, prominent nucleoli. The lymphoid blast cells sometimes have a so-called "hand mirror" shape, particularly if the malignancy is T cell-derived. Unlike myeloid blasts, lymphoid blasts tend not to be highly granulated.
Lymphocytosis
A common diagnostic problem involving lymphocytes that many physicians face at some point in their careers, regardless of specialty, is the evaluation of a patient with lymphocytosis, defined as an increase in absolute lymphocyte count, i.e., the percentage of lymphocytes multiplied by the total white blood cell (WBC) count. As we learned for myeloid cells, red cells, and platelets, the approach to lymphocytosis begins with categorization as either primary (due to a bone marrow or other primary hematologic disease) or secondary (due to a stimulus exogenous to the marrow). These categories can be distinguished on the basis of the peripheral blood smear, history, and sometimes flow cytometry. As an example, consider a college student who presents to urgent care clinic with two weeks of fever, profound malaise, a sore throat, and swollen cervical lymph nodes, who has a complete blood count (CBC) drawn that shows an elevated lymphocyte count, and whose blood chemistries reveal an increase in liver function tests. What would the diagnosis be if the following were seen on the smear?
Blood Smear of atypical lymphocyte
The answer is: infectious mononucleosis. The cell above is called an atypical lymphocyte, so-named because it has certain features of a lymphocyte (i.e., blue cytoplasm, absence of significant nuclear folding) but clearly looks atypical from a normal lymphocyte based on the large size of the cell and the "scalloping" of the plasma membrane against adjacent red blood cells. This is a hallmark of viral infections. In the context of a young patient with concomitant cervical lymphadenopathy, malaise, and transaminase elevation, EBV infection is the most likely cause.
Alternately, consider the case of an elderly man who is found to have an elevation in lymphocyte count, diffuse lymphadenopathy, and the following smear. What is the likely diagnosis?
Blood Smear
The answer is: chronic lymphocytic leukemia (CLL). This is a B cell lymphoproliferative disorder that is due to malignant B cells, which have a peculiar immunophenotype in that they express CD19 (a pan-B cell marker) and also CD5 (a T cell marker), and they are dim for CD20 (whereas most B cells have robust CD20 expression). The smear contains a preponderance of mature-appearing malignant lymphoid cells, along with large, irregular-appearing cells on the smear; these latter cells are called smudge cells and represent malignant lymphocytes that have been damaged by the smear preparation process. The constellation of asymptomatic lymphocytosis in an elderly person with diffuse lymphadenopathy and the above smear is very characteristic of CLL. Flow cytometry showing monoclonal CD19+ CD20(dim) CD5+ cells would support the diagnosis. [Of note, there are actually two B cell lymphomas that are CD19+ CD5+, one being CLL and the other being mantle cell lymphoma. The two can be distinguished partly on clinical grounds and also on the basis of CD23 expression, with CLL being CD23+ and mantle cell lymphoma being CD23-.]