Cell Growth Control
We all start as one cell and end up with several trillion. Because all cells arise from cells, cells must produce accurate and functional copies of themselves. Cell division produces two viable cells from a single cell. To maintain viability, each daughter cell must have a complete set of chromosomes and enough cellular machinery (organelles, proteins and other macromolecules) to sustain the essential biochemical pathways.

Cell Cycle Phases
The cell cycle is divided into 5 phases. G1 is a growth phase during which cells increase in size and eventually commit to cell division. S phase is when DNA replication occurs. G2 is another growth phase in which the centrosomes are duplicated. The centrosomes will set up the spindle that mediates separation of the chromosomes. Mitosis (M) has the most visible changes in cell structure. The mitotic spindle forms with microtubules emanating from the centrosomes and the chromosomes aligned in the center of the spindle. In the later stages of mitosis, the chromosomes migrate to opposite sides of the cell. Finally, during cytokinesis (C) the cell constricts in its middle, pinching together the cell membrane and leading to complete division into two cells. An additional stage is G0 which represents a quiescent state in which cells have exited the cell cycle and no longer divide. Most differentiated cells are in G0.

Cell Cycle Controllers: Cyclin and Cyclin-dependent Kinases
The phases of the cell cycle must proceed in an ordered fashion. DNA replication must precede chromosome separation, and chromosome separation must precede cytokinesis. A centralized controller ensures ordered progression through the phases of the cell cycle. The controller initiates cell cycle events at the proper time and makes sure that prior events are completed before the next stage is initiated. The cell cycle controller is a complex of two proteins: cyclin-dependent kinase (CDK) and cyclin. CDKs phosphorylate proteins to trigger specific cell cycle events. Cyclins regulate the activity of CDKs and help target them to their substrates.
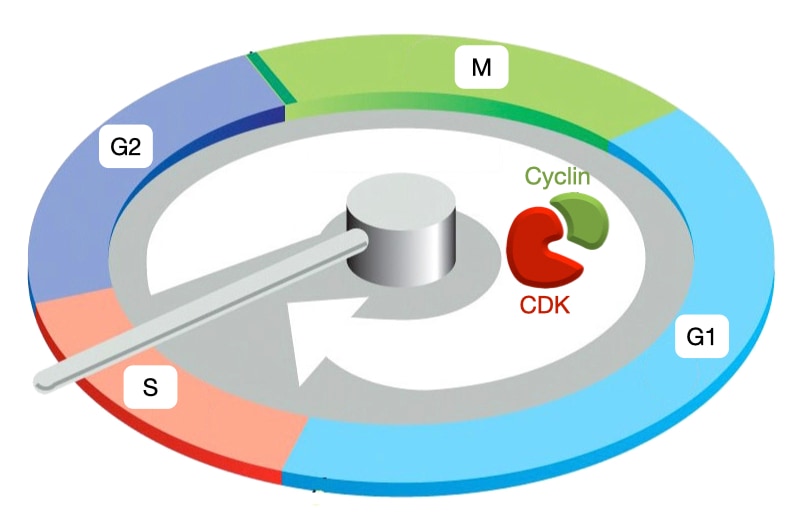
Cyclin Expression and Cell-cycle Phases
Different types of cyclin are expressed during different phases of cell cycle. When each cyclin is expressed, it associates with a cyclin-dependent kinase to trigger specific cell-cycle events. G1/S cyclins (cyclin E) trigger Start or entry into cell cycle. S phase cyclins (cyclin A) trigger DNA replication. M phase cyclins (cyclin B) initiate spindle assembly and attachment of microtubules to chromosomes. Lastly, the anaphase-promoting complex (APC) destroys all cyclins and initiates separation of sister chromatids. Cyclins activate CDKs by similar mechanism but lead CDKs to different targets.

Cyclin expression proceeds in a wave-like fashion through the cell cycle, and is how cyclins got their name: their levels cycle during cell division. Cyclin-CDK complexes in one phase trigger expression of cyclins in the next phase which ensures that events in the cell cycle proceed in an ordered fashion. In contrast, the amount of CDK in the cell remains at a consistent level throughout the cell cycle but CDK must associate with cyclin to be active.

Activation of Cyclin-CDK
The activity of CDKs occurs in three steps that involve binding to cyclin and post-translational phosphorylation.
The first step is CDK binding to cyclin. Cyclins bind to a conserved 100 amino acid domain in CDKs called the cyclin box and activate CDKs by partially opening its kinase pocket.
The second step is mediated by CDK-activating kinases (CAK). CAKs phosphorylate CDK leading to a complete opening of the substrate-binding site. CAKs seem to be constitutively active in most cells.
The third step is removal of an inhibitory phosphate from CDK. Wee1 is a kinase that adds a phosphate in ATP-binding pocket of CDKs which reduces kinase activity. CDK bound to cyclin and phosphorylated by Wee1 is inactive. Cdc25 is a phosphatase that removes the inhibitory phosphate from CDK to activate CDK-cyclin. This is the step at which checkpoints (see below) will often control progression into the next stage of the cell cycle.
The activation of cyclin-CDK is also regulated by positive feedback. Active cyclin-CDK phosphorylates Cdc25 to produce more active Cdc25 and as a result more active cyclin-CDK. Active cyclin-CDK also inhibits Wee1 reducing its ability to add the inhibitory phosphate to CDK.

Switch-like Activation of Cyclin-CDK
Why use a positive feedback loop? The positive feedback loop that activates scyclin-CDK commits the cell to each stage of the cell cycle. To demonstrate the importance of positive feedback in activating cyclin-CDK, we can compare what would happen if cyclin-CDK were activated without positive feedback. Recall that the amount of CDK in the cell is consistent throughout the cell cycle, but the amount of a specific cyclin increases before a stage of the cell cycle. When a signal triggers expression of a specific cyclin, the levels of that cyclin protein slowly increase, which, in the absence of positive feedback, lead to a linear increase in the amount of active cyclin-CDK.
There are two drawbacks to a slow, linear increase in cyclin-CDK activity. First, events in one stage of the cell cycle would likely start at different times as some events might require a higher concentration of active cyclin-CDK than other events. Second, the activity of cyclin-CDK is reversible because if the signal that triggered cyclin expression is removed, the amount of cyclin would decrease reducing the concentration of active cyclin-CDK.
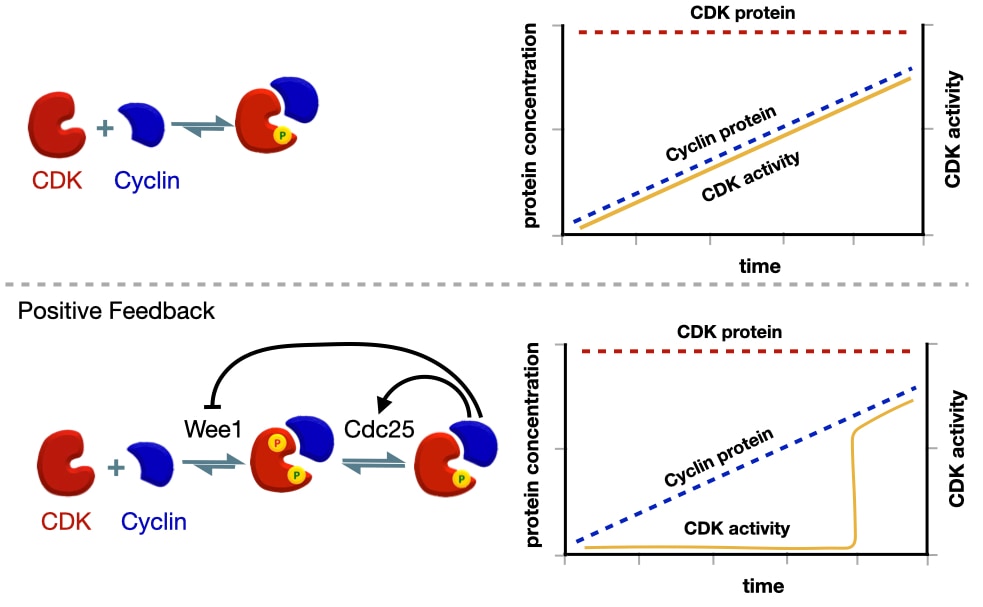
Positive feedback avoids these limitations by producing a switch-like activation of cyclin-CDK. Expression of cyclin starts before the next stage of the cell cycle. Cyclin binds CDK but remain inactive due to the inhibitory phosphate added by Wee1. This allows the cell to build up a large concentration of inactive cyclin-CDK before the next stage of the cell cycle. When the cell is ready to initiate the next stage of the cell cycle, it activates Ccd25 which removes the inhibitory phosphate on CDK leading to some active cyclin-CDK. Through the positive feedback loop, active cyclin-CDK activates more CDC25 and inhibits Wee1 producing even more active cyclin-CDK. The net effect is a sharp increase in the amount of active cyclin-CDK. In addition, the activity of cyclin-CDK now independent of the initial signal because cyclin-CDK is able to maintain its own activity through positive feedback.
Turning Off Cyclin-CDK
The use of positive feedback in the activation of cyclin-CDK means that active cyclin-CDK can't be turned off by simply removing the signal that triggered expression of the cyclin. To inactivate cyclin-CDK, cells target cyclins for destruction by ubiquitylation and then digestion via the proteosome.

Deciding When to Divide
The decision to enter the cell cycle is made at point in G1 called Start. After entering Start, a cell is committed to cell division because the cell commences chromosome replication and must finish the cell cycle or undergo apoptosis.
Because Start is the commitment step for the cell cycle, it is a point of intense regulation. Several factors determine whether a cell enters Start. Many single-celled organisms base their decision on whether to enter Start on the availability of extracellular nutrients. Our cells are usually surrounded by plenty of nutrients, and instead, they respond to signaling molecules to determine whether or not to divide. These signaling molecules are divided into two types. Mitogens are proteins or peptides that induce cells to start the cell cycle, whereas anti-mitogens inhibit entry into the cell cycle. The combination of mitogens and anti-mitogens allows tissues to regulate the pace of cell division and where in the tissue cells divide.

Start is triggered by the activation of G1/S cyclin-CDK which in mammalian cells is Cyclin E-Cdk2. Cyclin E-Cdk2 activates the S-phase complex, cyclin A-Cdk2, that initiates DNA replication. Because the activation of cyclin E-CDK commits the cell to divide, cells tightly control the activation of cyclin E-Cdk2. Cyclin E expression is controlled by another cyclin-CDK complex: cyclin D-Cdk4 (cyclin D also uses CDK6). Cyclin D levels increase throughout G1 and when the concentration of active cyclin D-Cdk4 reaches a certain level, the activation of cyclin E-Cdk2 is triggered and cells enter Start.
How Mitogens Trigger Cell Division
Mitogens bind receptors that trigger a signal transduction pathway that leads to the activation of cyclin D-Cdk4. Cyclin D-Cdk4 then triggers the activation of Cyclin E-Cdk2 which triggers entry into start and activation of Cyclin A-Cdk2. Cyclin A-Cdk2 initiate DNA replication.

How Mitogens Increase Expression of Cyclin D
Some mitogens, such as epidermal growth factor (EGF), increase the amount of cyclin D by activating transcription of the cyclin D gene. Mitogens bind receptors (often a receptor tyrosine kinase) that activates a guanine nucleotide exchange factor for the small GTP-binding protein Ras. Ras-GTP then activates a MAP kinase pathway that ultimately leads to the activation of a set of transcription factors including Myc. Myc increases the transcription of cyclin D. Because the receptor, Ras, MAP kinases, and Myc regulate the expression of cyclin D, mutations in the genes that encode these proteins that makes them continuously active can lead to uncontrolled cell division. For this reason the genes encoding the receptor, Ras, MAP kinases and Myc are know as oncogenes.

How Anti-mitogens Inhibit Entry into the Cell Cycle
Some anti-mitogens, such as TGF-β, inhibit the cell cycle by preventing cyclin D from binding Cdk4. Anti-mitogens binding a receptor (often a receptor serine/threonine kinase) that phosphorylates a protein called Smad (receptor-Smad). Upon phosphorylation, receptor-Smad dissociates from the receptor and binds its partner Smad4. The complex then enters the nucleus and increases the expression of a protein called Ink4. Ink4 binds Cdk4 and physically prevents it from binding Cyclin D. Because cyclin D cannot bind Cdk4, cyclin E is not expressed and the cell does not enter Start. Because INK4 inhibits the cell cycle, it is considered a tumor suppressor.

How Cyclin D-Cdk4 Activates Cyclin E-Cdk2
To generate cyclin E-CDK complexes, Cyclin D-Cdk4 increases the transcription cyclin E. Transcription of cyclin E is regulated by a set of transcription factors: E2F 1 - 5. E2F1, E2F2, E2F3 are activators of cyclin E transcription, and E2F4 and E2F5 are inhibitors of cyclin E transcription.

The interaction between the E2F proteins and the regulatory regions of the cyclin E gene is controlled by another protein called retinoblastoma protein or pRb. When E2F1, 2 or 3 are bound by pRb, they cannot associate with the enhancer of cyclin E, whereas when E2F4 or 5 is bound by pRb, both readily bind the repressor region of cyclin E to prevent transcription of cyclin E. Thus, in the presence of active pRb, cyclin E transcription is inhibited and cells do not divide. Because pRb inhibits cell division, it is called a tumor suppressor.

Cyclin D-Cdk4 triggers transcription of cyclin E by phosphorylating pRb. When phosphorylated, pRb cannot associate with the E2F proteins. Freed from pRb, E2F1, 2 or 3 can bind the enhancer of cyclin E to increase its transcription. In contrast, when E2F4 or 5 are not bound to pRb, they cannot bind the repressor of cyclin E.

Positive Feedback Loop to Activate Cyclin E-Cdk2
The activation of Cyclin E-Cdk2 is also controlled by a positive feedback loop. As mentioned above, cyclin D-Cdk4 phosphorylates pRb which leads to transcription of cyclin E and formation of cyclin E-Cdk2. Cyclin E-Cdk2 also phosphorylates pRb leading to more transcription of cyclin E. Once activated, cyclin E-Cdk2 can maintain its own activity through phosphorylation of pRb and sustained expression of cyclin E. At this point the activity of cyclin E-Cdk2 becomes independent of the mitogen. The mitogen is only needed to increase expression of cyclin D. Thus, for most cells, after a mitogen triggers entry into Start, the cell no longer needs the mitogen to proceed through the rest of the cell cycle.

Cell Growth and Cell Division
In addition to receiving a signal to initiate cell division, cells must accumulate enough macromolecules and organelles before entering the cell cycle. mTOR is a kinase and assembles with several other proteins to form mTOR complex (mTORC). mTORC plays a central role in integrating the nutrition status of the cell and activates pathways that increase production of protein and lipid. Specifically, mTORC phosphorylates proteins such as translation initiation factors and other activators of translation to increase production of protein. mTORC also activates transcription factors that induce expression of genes involved in lipid synthesis. Several key indicators of cell health and viability, including ATP concentration, oxygen levels and the concentration of amino acids, influences the activity of mTORC.

Certain mitogens activate signaling pathways that turn on mTORC to promote synthesis of protein and lipid to support cell division. The signaling pathway is similar to the one used by insulin to promote insertion of GLUT4 into the cell membrane. A mitogen binds to a tyrosine-kinase receptor, activating the receptor’s kinase domains and leading to cross-phosphorylation of the cytosolic domains of the receptor. The phosphorylated domains recruit scaffolding proteins which in turn bind PI-3 kinase. PI-3 kinase converts phosphatidylinositol-2-phosphate (PIP2) into phosphatidlyinositol-3-phosphate (PIP3). PIP3 recruits PDK1 and AKT to the membrane. PDK1 phosphorylates and activates AKT1. AKT1 activates mTORC by keeping the GTP-binding protein rheb in a GTP-bound state. Active mTORC increase production of protein and lipid as described above. Thus, mitogens activate pathways that stimulate the cell cycle and increase the production of key macromolecules for cell growth.

Cell-cycle Checkpoints
The cell monitors its progress through the cell cycle to ensure each event is complete before proceeding to next step and checks that conditions are okay before proceeding to the next step. Checkpoints sense unreplicated DNA, DNA damage and unattached chromosomes during mitosis. When activated, checkpoints arrest the cell cycle before the next stage. Activation of checkpoint inhibits the activity of cyclin-CDK complexes and prevents them from initiating next phase of cell cycle.

DNA Damage Checkpoint
As mentioned, the cell monitors the state of its DNA and arrests the cell cycle if it detects damaged or unreplicated DNA. DNA damage arrests the cell cycle through two mechanisms.
DNA Damage and P53
DNA damage leads to the activation of a transcription factor called p53. Cells continually express p53 but most p53 is associated with another protein called Mdm2. Mdm2 prevents p53 from entering the nucleus to activate gene expression and it also leads to the ubiquitylation of p53 which targets p53 for digestion by the proteosome. Thus, Mdm2 keeps p53 concentrations low and prevents it from activating transcription.
DNA damage is recognized by a set of kinases (ATM/ATR and Chk1/Chk2). These kinases phosphorylate Mdm2 causing it to dissociate from p53. Released from Mdm2, p53 can enter the nucleus where it activates the transcription of genes which encode proteins that arrest the cell cycle. One of these proteins is p21. P21 binds Cyclin-CDK complexes, such as cyclin E-Cdk2, and inhibits their activity to arrest the cell cycle. Because p53 and p21 inhibit the cell cycle, they are known as tumor suppressors.

DNA damage also arrests the the cell cycle by reducing the amount of Cdc25. Recall that Cdc25 is a phosphatase which removes the inhibitory phosphate from Cdk2 and leads to its full activation. Kinases activated by DNA damage phosphorylate Cdc25 which exposes a degradation motif. Cdc25 is ubiquitylated and then digested by the proteosome.

p53 and the Inhibition of Oncogenesis
As mentioned above, many components in the mitogen-activated signal transduction pathways can trigger uncontrolled cell division when mutated. These components include Ras, Myc and the E2F proteins that increase transcription of cyclin D. The genes that encodes these proteins are known as oncogenes. Mutations in these genes can produce proteins that are constitutively active or overexpressed leading to constant expression of cyclin D, even in the absence of a mitogen. This allows cells to enter start Start independently of mitogens and prevents tissues from regulating the proliferation of its cells. Cell division independent of mitogen is one of the hallmarks of cancer.
Fortunately, mutations in Ras and Myc also lead to the activation of p53. Besides increasing the transcription of cyclin D, the oncogenes also increase the transcription of p14-ARF. p14-ARF binds Mdm2 and prevents it from interacting with p53. Consequently, p53 enters the nucleus and activates transcription of a gene that encodes p21. p21 binds cyclin-CDK complexes and inhibits their activity to prevent progress through the cell cycle. Because p21 is slows the cell cycle, p21 is a tumor suppressor.

The finding that p53 can prevent cell division even when a cell has an oncogenic mutation in Ras or Myc led to the two-hit model of tumorigenesis. In this model, a cell becomes cancerous only after it receives two mutations: one in an oncogene (e.g. Ras, Myc) and the other in a tumor suppressor (e.g. p53, p21, Ink4). Most tumors have mutations in several different genes each of which contributes to the rapid cell division independent of mitogens.
p53 and Apoptosis
In addition to arresting the cell cycle, P53 can also induce apoptosis in cells. When activated, P53 can increase the transcription of gene called Bax. Bax is cytosolic protein that under certain conditions translocates to the outer membrane of mitochondria. There, it assembles into a channel that allows cytochrome c to move from the mitochondria to the cytosol. In the cytosol cytochrome c can activate the caspases that initiate apoptosis.
Chromosome Segregation and the Completion of Mitosis
Mitosis is the phase of the cell cycle during which the cell separates its chromosomes equally into the two daughter cells. The different stages of mitosis can be resolved by light microscopy and the major events in the each stage are well defined.

Prophase sees the condensation of the chromosomes and separation of the two centrosomes to opposite sides of the cell. Centrosomes nucleate microtubule growth which will form the spindle that ultimately separates the chromosomes.
Prometaphase includes the breakdown of the nuclear envelope which exposes the chromosomes to the cytosol including microtubules from the centrosomes.
Metaphases reveals the mitotic spindle with chromosomes in the center of the cell and centrosomes on opposite sides. Microtubules radiate from the centrosomes and connect to the chromosomes.
Ananphase is the initial step in separating the chromosomes and is divided into two phases. Anaphase A sees the chromosomes starting to pull apart from each other which is driven by depolymerization of the microtubules that are attached to the chromosomes. Anaphase B includes an elongation of the spindle as the two centrosomes move away from each other. Motor proteins attached to the microtubules generate the force to push apart the centrosomes.
Telophase shows the cell membrane starting to pinch together in the middle of the cell and reformation of the nuclear envelope.
Cytokinesis completes cell division as the cell membrane at the middle of the cell completely pinches together and eventually fuses to generate two separate cells. Actin and myosin filaments constrict the cell membrane during cytokinesis.
Mitotic Spindle
To accurately segregate the chromosomes, cells build a spindle of microtubules during metaphase. Three types of microtubules grow from the centrosomes to form the spindle. Astral microtubules extend from the centrosomes away from the center of the cell and toward the cell membrane. Interpolar microtubules extend toward the center of the cell and overlap with interpolar microtubules coming from the opposite centrosome. Astral and interpolar microtubules, along with the motor proteins that bind to them, position the spindle in the center of the cell and will elongate the spindle during anaphase B to help separate the chromosomes. Kinetochore microtubules attach to chromosomes at the centromere region. Kinetochore microtubules will generate the force to separate the chromosomes during anaphase A.

Mitotic Checkpoint
The other major cell cycle checkpoint occurs during mitosis when the cell is segregating the chromosomes into each future daughter cell. Producing two viable daughter cells requires that each cell receives a complete set of the chromosomes. Improper segregation of the chromosomes can lead to tumorigenesis or developmental diseases, such as Down's syndrome.
Prior to initiating anaphase and separation of the chromosomes, cells check that all of the chromosomes are properly attached to microtubules in the spindle. If even one chromosome is not attached correctly to microtubules in the spindle, the cell will remain arrested in metaphase. This is the mitotic checkpoint and it regulates the transition from metaphase to anaphase.

Two major mechanisms arrest cells in metaphase and prevent premature separation of chromosomes. The first is active cyclin B-CDK2. CDK2 kinase is highly active through metaphase but its activity plunges during the transition to anaphase. Second, a protein complex called cohesin wraps the sister chromatids and physically prevents their separation. Thus, the transition to anaphase and separation of chromosomes requires inactivation of CDK2 and removal of cohesin from chromosomes.

The anaphase promoting complex (APC) reduces CDK2 activity and triggers the destruction of cohesin. APC is an ubiquitin ligase which is inactive until it binds its partner Cdc20. A single, unattached chromosome is sufficient to keep cytosolic Cdc20 levels low. Only when all chromosomes are properly attached to the spindle do Cdc20 levels increase and bind APC.

APC bound to Cdc20 has two main targets. First, it ubiquitylates Cyclin B which triggers its digestion in the proteasome. The loss of cyclin B reduces Cdk2 activity and is one of the steps required for the cell to transition from metaphase to anaphase. Second, APC ubiquitylates securin which is bound to an enzyme called separase. Ubiquitylation of securin triggers its digestion in the proteasome, freeing and activating separase.

Active separase digests cohesin proteins that tether the sister chromatids. Once cohesin has been removed, tension generated by the microtubules in the spindle can now begin to separate the chromosomes and the cells transitions into anaphase to complete mitosis.
